Sie ist eine der seltensten und tödlichsten Krankheiten der Welt. GM1 Gangliosidose. Weltweit leiden ungefähr 6.000 Kinder an dieser Erkrankung. Auch die Mönchengladbacherin Mila Kapturczak erhielt erst im zarten Alter von knapp 5 Jahren die tödliche Diagnose.
Bitte fügen Sie Ihrer Spende die Mitteilung „Mila“ hinzu.
Alternativ können Sie auch per Überweisung spenden:
Empfänger: Löwenkinder Viersen
Verwendungszweck: MILA
Konto: Sparkasse Krefeld
IBAN: DE65 3205 0000 0000 4708 80
Sie erhalten eine Spendenquittung
Mila Kapturczak
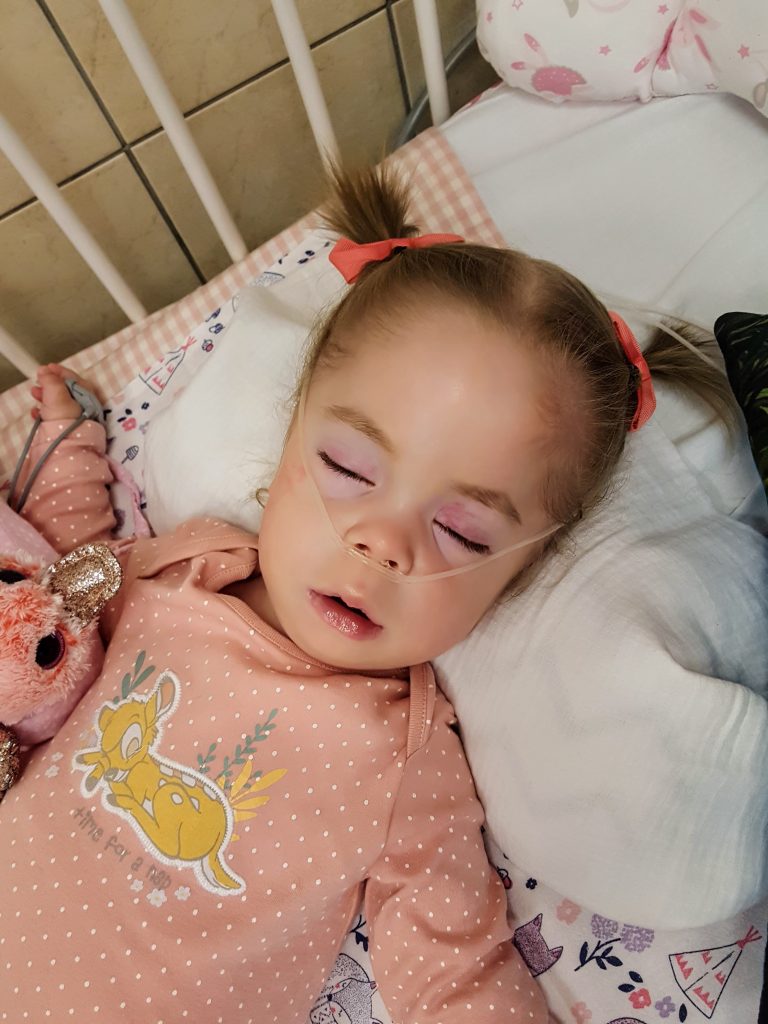
Mila wurde am 27.April 2014 als ganz normales Kind in Mönchengladbach geboren. Sie wuchs gesund auf und entwickelte sich in den ersten drei Lebensjahren prächtig. Nichts deutete auf die tödliche Erkrankung hin. Kurz nach ihrem dritten Geburtstag veränderte sich Mila. Die Veränderung zeigte sich in Form von Sprachschwierigkeiten und setzte sich mit zunehmenden Lauf- und Bewegungseinschränkungen fort. Auch ihre koordinativen Fähigkeiten nahmen von Tag zu Tag ab.
Die Eltern konsultierten ihren Kinderarzt in Jüchen, der aber zunächst nur die Symptome behandeln konnte und Mila zusätzlich an eine Kinder-Reha-Klinik in Meerbusch verwies. Dort wurden die krankheitsbedingten Begleitmaßnahmen durch Therapien verbessert. Darüber hinaus halfen dem jungen Mädchen auch die symptomatisch eingesetzten Medikamente gegen ihre zahlreichen Krampfanfälle nicht weiter.
Die Diagnose
Am 28.März 2019 war es soweit. Mila erhielt ihre eindeutige Diagnose in der Düsseldorfer Uniklinik. GM1 Gangliosidose 1 Typ 2, bekannt als Derry-Syndrom. Es war zugleich ihr Todesurteil. Aus Erfahrungsberichten der Vergangenheit weiß man, dass Kinder mit dieser Erkrankung eine Lebenserwartung von maximal 10-11 Jahren haben.


Die Krankheit
Bei der GM1 Gangliosidose handelt es sich um eine erblich bedingte, sehr seltene lysosomale, d.h. in den Zellen stattfindende Speicherkrankheit. Es ist der Mangel an dem Enzym Beta-Galaktosidase. Dieses Enzym kommt in den Zellen des Nervensystems vor und ist dort für den Abbau und damit für die Entgiftung von 3 wesentlichen toxischen Substanzen verantwortlich, den sog. Gangliosiden.
Bei den Kindern zeigt sich das Krankheitsbild in Form von Viszeromegalie, d.h. abnormale Vergrößerung der inneren Organe, Skelettdysplasie, d.h. Beeinträchtigung des Knochen- und Knorpelwachstums, Herzproblemen und zahlreichen neurologischen Beeinträchtigungen, die sich in den verschiedensten Ausprägungen manifestieren.
Diese Diagnose veränderte Milas Leben und das der Eltern kolossal. Doch aufgeben war keine Option.
Der Vater wandte sich zunächst hilflos an den Apotheker Oliver Dienst. Zusammen machten sich beide auf die Suche und Recherche über diese Erkrankung.
Verbreitung auf der Welt
Die größte Zahl von den über 6.000 auf der Welt lebenden Gangliosidose (GM1)-Patienten:innen finden sich in Indien mit 2740 und in Brasilien mit rund 2030 Kindern, während in Europa etwa über 500 Kinder von der seltenen Krankheit betroffen sind. Die Dunkelziffer ist vermutlich um ein Vielfaches höher.
Wettlauf gegen die Zeit
Der Vater und der Apotheker erarbeiteten sich in kürzester Zeit ein weltweites Netzwerk und stießen wie durch Zufall auf den Biochemiker Dr. Axel Heinemann, der seine Recherche über sog. medizinische Datenbanken noch stärker intensivierte.
So wurde in den 80er Jahren in Japan mit einer Substanz geforscht, die mit Morbus Gaucher in Zusammenhang gebracht wurde. Auch beim Morbus Gaucher ist, ähnlich wie bei der GM1 Gangliosidose, eine lysosomale Speicherkrankheit die Ursache. Auch hier wird durch eine genetisch bedingte Störung der Abbau von ähnlich toxischen Substanzen verhindert.


Der Wirkstoff
Aloxistatin heißt die Substanz und wurde von einem Schweizer Unternehmen im Jahr 2015 patentiert und an diversen Speicherkrankheiten eingesetzt. Der erfolgversprechendste Einsatz erfolgte an einem ukrainischen Kind im Jahr 2018. Die Beta-Galaktoidase-Aktivität konnte bei diesem Kind gesteigert werden. Doch leider kam der Wirkstoff zu spät. Das ukrainische Kind verstarb kurze Zeit später an den Folgen der heimtückischen Krankheit. Die weitere Verfolgung, die Substanz weiter bei an GM1 Gangliosidose erkrankten Kindern einzusetzen, „versandete“ nahezu.
Erst durch die Recherche des Vaters, des Biochemikers und des Apothekers wurde die Substanz wieder „zum Leben“ erweckt. Nun gab es vielleicht eine hoffnungsbringende Substanz, aber noch lange kein Arzneimittel. Und Richtlinien in Deutschland schreiben klar vor, was die Gabe von nicht zugelassenen Arzneimitteln anbelangt. Auf deutsch: Man kann einem Kind nicht irgendeine Substanz, an der jemand irgendwo auf der Welt geforscht hat, geben und ausprobieren. Vor allem eine „Giftigkeit“ muss durch viele Studien ausgeschlossen sein. Und die Frage: woher beziehen und wie finanzieren?
Die OFF-Label-Studie
Zusammen mit der Uniklinik Gießen, und der Kinderärztin Dr. Christina Lampe, die sich mit vorwiegend seltenen Erkrankungen befasst und die Krankheit „GM1 Gangliosidose“ nicht nur kannte, sondern auch Erfahrungen in der Vergangenheit damit gemacht hatte, bot sich als begleitendes Institut für eine mögliche Therapie bei Mila an.
Im Rahmen einer Off-Label-Studie sollte die Therapie bei Mila durch die Krankenkasse bewilligt werden.
Der Start der Therapie
Zu Beginn wurde die Substanz noch aus der Hauptstadt Estlands, Tallinn, bezogen. 50g zum Preis von 100.000 Euro. Und wie gesagt, mit der Substanz hatten wir noch lange kein Arzneimittel. Also, wie sollte Mila die Substanz zukünftig einnehmen? Schlucken? Trinken? Als Zäpfchen? Inhalieren? Auf der Suche nach der geeigneten galenischen Möglichkeit entschieden wir uns für die Kapselherstellung. Und wieviel von dem „teuren Pulver“ sollte in jede Kapsel hinein? Wie hoch ist die Dosis bei einem Kind? Denn weder die Applikationsform, noch die Dosis waren bekannt. Also ausprobieren und testen. Auf die Hilfe von Mila angewiesen, starteten wir mit der Gabe langsam steigernd.


Ein Hoffnungsschimmer
Nach einer mehrwöchigen Gabe der Substanz passierte folgendes. Bei Mila, die sich zum Zeitpunkt der ersten Gabe in einem sehr schlechten Zustand mit Gehverlust, starken motorischen Problemen, Ataxie, Krampfanfällen, Sprachverlust, Muskelhypotonie befand, stieg die bedeutsame Aktivität der Beta-Galaktosidase in Gegenwart von Aloxistatin in den Fibroblasten (Zellen des Bindegewebes) signifikant an. Mila nahm von nun an 3 x täglich 50mg mit dem Essen ein. Die Kapseln wurden vor der Anwendung geöffnet und das Pulver „pur“ gegeben.
Nach nunmehr 6 Monaten dieser „Behandlung“ ist Mila aufmerksamer, konzentrierter und wesentlich fitter. Sie lächelt seitdem wieder von morgen bis abends. Insbesondere ist es ein Riesen Erleichterung für die Eltern, dass nach der Einnahme von Aloxistatin Mila wieder deutlich darauf aufmerksam macht, dass Sie auf die Toilette muss.
Befürchtete unbekannte Nebenwirkungen blieben aus.
Das zweite Kind Biborkla
Was sich wie ein Wunder etablierte, sollte nun an einem weiteren 18 Monate altem Kind aus Ungarn getestet werden. Das ungarische Mädchen Biborkla befand sich zum Zeitpunkt der ersten Gabe in einem sehr ernsten Zustand mit allgemeinen Ödemen. Sie war nicht in der Lage den Kopf und Gliedmaßen zu bewegen.
Sie litt an Krampfanfällen und wurde mit 0,5 – 1 Liter Sauerstoff pro Minute künstlich beatmet. Sie litt an Herz-, Atemwegs- und Verdauungsproblemen mit Ausscheidungsstörungen.
Seit Mai 2022 wird die junge, schwer kranke Patientin mit Aloxistatin behandelt und zeigt wie Mila bereits nach kurzer Zeit Besserung. Die Anzahl der Anfälle reduzierten sich extrem, die Ödeme resorbierten sich vollständig, die Sauerstoffversorgung reduzierte sich auf 0,25 – 0,5 Liter pro Minute und ihre Urinmenge nahm zu.
Ihr Magen verkleinerte sich und sie begann selbständig ihren Kopf und ihre Gliedmaßen zu bewegen. Mittlerweile benötigt sie keinen Sauerstoff mehr und kann ihre Eltern wieder anlächeln. Auch hier sind unerwartete Nebenwirkungen bis heute ausgeblieben.
Projektskizze
Entwicklung einer Aloxistatin-basierten
oralen Applikationsform mit Hilfe von
Einschlussverbindungen
Nun ist es an der Zeit noch vielen weiteren Kindern auf dieser Welt das Leben zu retten.
In diesem Entwicklungsprojekt wird versucht, eine Aloxistatin-haltige, orale Darreichungsform auf Basis eines Rezepturarzneimittels zu entwickeln. Zur Verbesserung der Resorptionsquote und daraus resultierender erhöhter Bioverfügbarkeit steht die Formulierung einer Einschlussverbindung von Aloxistatin mit einem adäquaten Komplexbildner im Mittelpunkt des gemeinsamen Vorhabens.
Dabei sollen insbesondere folgende Ziele verfolgt werden:
• Identifizierung eines geeigneten Komplexbildners für die Herstellung einer hydrophilen Einschlussverbindung von Aloxistatin
• Ermittlung der optimalen Substratlöslichkeit in Wasser
• Löslichkeitsprüfung durch UV-Vis-Spektroskopie (UV-Vis) und/oder Hochleistungsflüssigkeitschromatographie (HPLC) und Validierung der Verfahren
• Qualitative Charakterisierung der Aloxistatin-Einschlussverbindung mittels FT-IR Spektroskopie und dynamische Differenzkaloriemetrie (DDK, engl. differential scanning calorimetry, DSC) sowie ggf. durch die Anwendung weiterer spektroskopischer Verfahren (z.B. 1H-NMR Techniken)
• Galenische Entwicklung einer oralen Darreichungsform
• Generierung von Stabilitätsdaten der oralen Applikationsform und Gesamtkeimzahlbestimmung (TAMC), ggf. Konservierungsmittelbelastungstest
Knapp 1 Millionen Euro hat der Vater Lothar Möller bereits gesammelt. Für Investoren nicht nur eine Herzensangelegenheit, sondern vielleicht irgendwann auch ein wirtschaftlicher Erfolg. Denn der zu bedienende Markt ist groß. Auch wenn 6.000 Kinder zunächst nur ein Anfang sind, ist dieser Anfang jeden Cent wert.
Aber eins ist sicher. Die Zeit ist gegen Mila und die anderen Kinder.
Bitte fügen Sie Ihrer Spende die Mitteilung „Mila“ hinzu.
Alternativ können Sie auch per Überweisung spenden:
Empfänger: Löwenkinder Viersen
Verwendungszweck: MILA
Konto: Sparkasse Krefeld
IBAN: DE65 3205 0000 0000 4708 80
Sie erhalten eine Spendenquittung
Projektablauf und Konzept
Doch hierzu bedarf es an Geldern. Die voraussichtlichen Kosten betragen etwa 250.000 €
AP 1. Identifizierung eines geeigneten Komplexbildners zur Komplexierung von Aloxistatin
- Vorversuche mit unterschiedlichen Komplexbildnern
- Ermittlung eines geeigneten Verfahrens zur Komplexierung
AP 2. Entwicklung einer stabilen Lösung der Einschlussverbindung
- Entwicklung einer stabilen Lösung mit der Aloxistatin-Einschlussverbindung in einer ausreichenden, therapeutisch sinnvollen Konzentration
AP 3. Löslichkeitsprüfung durch spektroskopische Verfahren
- Vorversuche zur Ermittlung eines geeigneten Verfahrens zur Identifizierung und Quantifizierung der Aloxistatin-Einschlussverbindung mittels UV-Vis-Spektroskopie (UV-Vis) und/oder Hochleistungsflüssigkeitschromatographie (HPLC)
- Entwicklung und Validierung des spektroskopischen und/oder chromatographischen Analyseverfahrens
AP 4. Qualitative Charakterisierung der Aloxistatin-Einschlussverbindung
- Qualitative Charakterisierung der Aloxistatin-Einschlussverbindung mittels FT-IR Spektroskopie und dynamische Differenzkaloriemetrie (DDK, engl. differential scanning calorimetry, DSC) sowie ggf. weiterer spektroskopischer Verfahren (z.B. 1H-NMR Spektroskopie)
AP 5. galenische Entwicklung der oralen Darreichungsform
- Galenische Formulierungsentwicklung zu einer applikationsfähigen Darreichungsform
AP 6. Methodenvalidierung für die Stabilitätsprüfung
- Entwicklung und Validierung einer chromatographischen und/oder spektroskopischen Analysemethode zur Stabilitätsprüfung der oralen Formulierung
AP 7. Generierung von Stabilitätsdaten der oralen Formulierung
- Einlagerung bei unterschiedlichen Klimazonen (25°C und 40°C) und Testung auf Stabilität und Packmittelkompatibilität
- Gesamtkeimzahlbestimmung (Total Aerobic Microbial Count (TAMC)) – am Anfang und Ende der Stabilitätslaufzeit
- Ggf. Durchführung eines vorgeschalteten „Tests auf ausreichende Konservierung“ im Falle der Formulierung einer flüssigen Zubereitung